



The transition metal catalyzed polymerizaion of olefins can be thought of as a reaction in which the carbon-carbon π-bond in α-olefins (1-alkenes) are cut and then remade as a carbon-carbon σ-bonds, linking the olefins together in long hydrocarbon chains. This reaction is exothermic by ~ 20 Kcal/mol, the energy difference between the carbon-carbon π- and σ-bonds. For typical early transition metal, d0 catalysts, the activation energy is ~ 10 Kcal/mole, making this a very facile reaction.
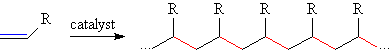
These long chain macromolecules are known as polymers (from the Greek, poly = many and mer = units) and are the principal constituents of many common plastics. Some familar plastics made via early transition metal catalyzed polymerization include: high density polyethylene (HDPE, R=H), linear low density polyethylene (LLDPE, R = mostly H with some Et, Bu or Hx), polypropylene (R = Me) and ethylene-propylene-diene-modified rubber (EPDM, R = H, Me and Alkenyl).
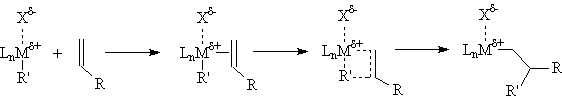
The commonly accepted mechanism for the olefin polymerization reaction is shown below. A severely electron deficient metal center coordinates the π-bond of an olefin to form a weakly bound olefin complex; the bond is weak because there are no d-electrons to form a π-bond. The bound olefin then inserts into a metal-carbon bond within the complex via a four center transition state, forming new metal-carbon and carbon-carbon bonds. This process then repeats, extending the chain indefinitely.
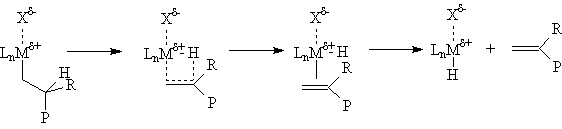
Chain growth can be terminated by a number of processes. One of the most impotant is β-hydride elimination. Since this is simply the reverse of insertion, the activation energy is ~ 30 Kcal/mole plus the energy difference between the metal-carbon and metal-hydrogen bonds. Catalysts with relatively strong M-C bonds give higher molecular weight polymers, while those with relatively strong M-H bonds give lower molecular weight polymers (waxes). Beta-hydride elimination leads to polymers with vinyl (R=H) or vinylidene (R=Me, Et, Pr...) end groups. The vinyl terminated polymers are, of course, α-olefins and may also be incorporated into the growing polymer chains giving "long chain branched" products under appropriate conditions.
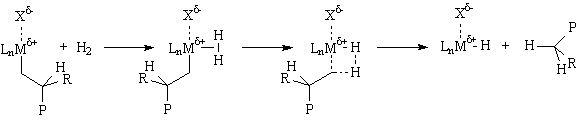
Another termination process of great practical use is hydrogenolysis. This sigma-bond metathesis reaction leads to polymers with saturated end groups:
There have been many generations of olefin polymerization catalysts:
While these catalysts are exceedingly active, they have an exceedingly low tolerance for functional groups because of their Lewis acidic nature. Typically they contain multiple active sites with varying, but generally poor, reactivity ratios for α-olefins relative to ethylene. Little is known about the nature of the actual catalytic species in these systems.
This class of catalysts came to include rationally designed systems, such as Ewen's Et(Ind)2ZrCl2/MAO (isotactic polypropylene: Ewen, J. A., J. Am. Chem Soc. 1984, 106, 6355-6364), and iPr(Cp)(Flr)ZrCl2/MAO (syndiotactic polypropylene: Ewen, J. A.; Jones, R. L.; Razavi, A.; Ferrara, J. D., J. Am. Chem. Soc. 1988, 110, 6255-6256). These catalysts rejuvenated olefin polymerization catalysis research in the early 1980's. Despite a quarter century of intense academic and industrial research, the nature of MAO is still largly unknown. Active site counting studies and chromatographic analysis suggest that the MAO exists as clusters with ~10-15 aluminum atoms and acts as both a methyl transfer agent and a Lewis acid to form anions which are weakly coordinating to the cationic metal methyl complexes which are the active catalysts.
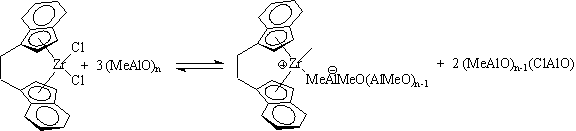
These catalysts have good activity and remarkable tunability via ligand changes in the pre-catalyst complex. For example, the chiral active site of the Kaminsky-Brintzinger catalyst system (Kaminsky, W.; Kulper, K.; Brintzinger, H.H.; Wild, F.R.W.P. Angew. Chem. Int. Ed. Eng.1985, 24, 507.) depicted above generates isotactic polypropylene. However, the use of large amounts of MAO (~ 1000 equivalents, presumably to drive the above equilibrium to the right) results in high catalyst cost and catalyst residue loading of the polymer.
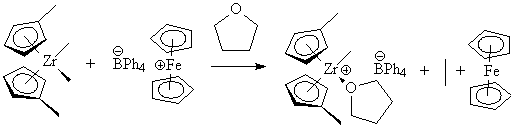
Like the Kaminsky type catalysts, the Jordan type catalysts have exceptional tunability via the metal ligands. The early versions of these catalysts showed only modest activity, due to competition for the active site between the olefin and the ether.
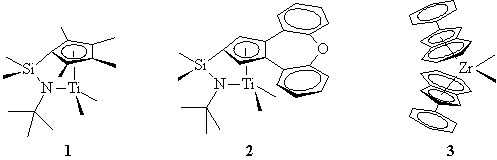
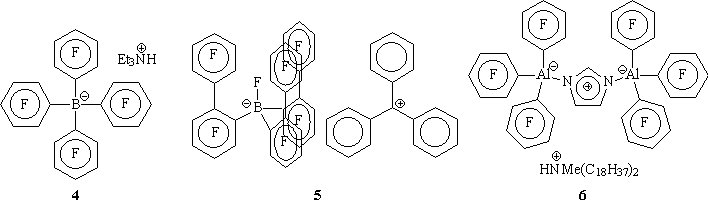
1The author and Rob Toreki acknowledge Larry Jones of DSM PTG for his insightful editorial comments and the reference list provided here.