



A fluxional molecule is one that undergoes a dynamic molecular process that interchanges two or more chemically and/or magnetically different groups a molecule. If the rate of this exchange is faster than the time scale of our spectroscopic observation, these two different groups can appear to be identical. We also use the term, dynamic exchange process, to express a molecular motion that interchanges the positions of the inequivalent groups.
Multinuclear NMR spectroscopy, one of the favorite tools of the organometallic chemist, is one of the most common ways of observing dynamic behavior.
Classical kinetics can often be used to determine the rate constant and activation energy of a chemical reaction. In a typical study, changes in concentration of products and/or reactants versus time are monitored using any number of experimental techniques (IR, NMR and UV-VIS are the most common). This method works well for reactions that take place on the laboratory time scale (minutes to hours) where the rate constants for the reactions are typically 10-6 to 10-3 s-1.
This analysis becomes more complicated when we have to consider reversible reactions or systems that are at equilibrium. For example:
The prototypical example of such a system is the axial-equatorial interconversion of the chair form of cyclohexane. At room temperature the axial and equatorial protons are interchanged by a dynamic (fluxional) process in which the ring undergoes a "chair-chair" conformation change. As shown here, Ha and Hb are interchanged between axial and equatorial positions:
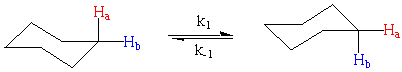
To understand why this complicates our analysis remember that in the 1H NMR experiment we irradiate the protons to flip their nuclear spins and then wait as they give off this excess energy. The energy (frequency) of this relaxation is what we more commonly call the chemical shift of our proton. It takes time for our protons to relax to their nuclear ground states and this relaxation is governed by both the spin-lattice, T1, and spin-spin, T2, relaxation time.
Imagine that we irradiate a proton while it is in the equatorial position. Under normal circumstances, the proton would relax and we would detect it at a chemical shift characteristic of equatorial protons. However, if the molecule rearranges so that this proton is in the axial position when it relaxes, the chemical shift would be consistent with an axial proton.
To prevent exchange from occurring, all we have to do is cool the sample to a sufficiently low temperature. At -90 degrees C, the axial and equatorial protons of cyclohexane no longer interchange and are resolved as two separate resonances. But as we raise the temperature, the two peaks move together and broaden, indicating that there is some exchange, a regime we call slow exchange.
When the two peaks merge such that there is no distinguishable valley between them we say that the peaks have coalesced. As we raise the temperature even more, the merged broad peak sharpens again. At this point, the lifetime of a species as axial or equatorial is much shorter than the time scale of the experiment (flipping the nuclear spin and observing the relaxation). We call such a system fast on the NMR timescale or denote it as being at the high temperature limit.
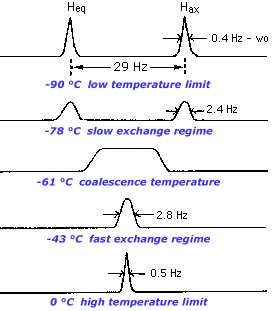
Figure 1. VT-NMR spectrum of cyclohexane-d11 (See Kegley, page 21). Note: All peaks are singlets instead of doublets because JH-D is small and 11 of the 12 protons are deuterated. In a non-deuterated sample, each peak would be a doublet with a Jax-eq of approximately 13 Hz.
An organometallic system that undergoes a similar interconversion, but at higher temperature is the bent metallocene complex, Cp2TiS5. In the structures below, notice that Sa and Se of the pentasulfide unit are closer to one cyclopentadienyl ring than the other. This creates two chemically and magnetically inequivalent Cp rings which appear as separate signals in the 1H or 13C NMR spectrum.
Above room temperature, Cp2TiS5 undergoes a chair-chair rearrangement which effectively switches the polysulfide ligand from one side of the molecule to the other. As far as our NMR spectrometer is concerned, the two Cp rings (conveniently marked in blue and red) appear to have exchanged positions even though they did not actually move. The sulfurs have been marked to show you that this process is not merely rotation of the entire molecule by 180 degrees, but an inversion of the ring:
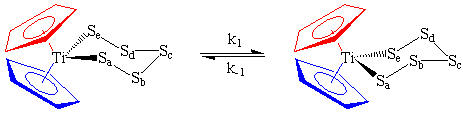
| Note: The Cp rings themselves freely rotate about the Ti-ring centroid with an exceedingly low energy barrier. Therefore, all five protons on the same ring act chemically equivalent at all temperatures and appear as a singlet even though some may be closer to the S5 ring in the static picture shown above. |
This chair-chair inversion exchange process is slow at room temperature, but can be conveniently studied over the range 20-120 degrees C using variable temperature 1H NMR. This makes a good advanced undergraduate laboratory experiment which you can get at http://www.chem.uky.edu/courses/che450g/handouts/cp2tis5.html if they are still using the experiment in the lab course I used to teach.
Using methods described in the references below, an Arrhenius and Eyring plot can be obtained from variable temperature NMR data. This permits one to calculate the activation energy, Ea, as well as the  G, H and S of activation for the dynamic process. This, in turn, can give you valuable information that you can use to support or rule out certain mechanisms. We hope to include this information in this document in the near future.
G, H and S of activation for the dynamic process. This, in turn, can give you valuable information that you can use to support or rule out certain mechanisms. We hope to include this information in this document in the near future.
This is another useful technique; watch for a separate entry sometime in the near future.
The following problems involve examples of dynamic behavior. These problems are mechanistic in nature so you will not be presented with multiple choice answers here. Instead, print the problem out, find a solution that fits the data and then come back to this page.
When you hit the "Start Solving" button, you will be presented with a series of questions that will probe your assessment. Each will either allow you to proceed or point out an error in your reasoning. If you do not get a correct answer along the way then stop, examine the feedback and think about the problem some more before continuing. Remember, these problems are not meant to be solved in five minutes.
THESE EXERCISES ARE NOT CURRENTLY WORKING - We moved to a new server platform in September 2025 and I have to go back and redo the coding that drives the grading. Stay tuned...